
The European Medical Device Regulation (EU MDR) will greatly affect medical device manufacturers operating in the European Union (EU). The regulation entered into application in May 2021 with transition periods based on device classification.
The EU MDR has introduced significant challenges for the medical device industry. In November 2022, Politico reported, “The new rules have inadvertently triggered a new public health emergency—a looming cliff edge in unavailable devices.”
The EU MDR is impacting both companies and patients, with some medical devices already being removed from the market, awaiting certification.
So, as a medical device manufacturer, what do you need to know? This article answers the questions:
- What is the European Medical Device Regulation?
- How are medical devices classified under EU MDR?
- When are the transition period and compliance deadlines?
- What are the impacts of the European MDR?
- What is the role of notified bodies?
- How can medical device manufacturers prepare?
European Medical Device Regulation: 5 Steps to Prepare for What’s Coming.
This guide provides a detailed checklist to help you get ready.
What is the European Medical Device Regulation?
The EU MDR is a set of rules for medical devices in the EU. It replaces the Medical Device Directive (MDD) and Active Implantable Medical Devices Directive (AIMDD). The regulation, in full application pending transition periods since May 2021, aims to keep EU citizens safe when using medical devices.
The European MDR is a major overhaul of the existing regulatory framework for medical devices in Europe.
The European Commission says, “The new regulations create a robust, transparent, and sustainable regulatory framework, recognised internationally, improving clinical safety and creating fair market access for manufacturers and healthcare professionals.”
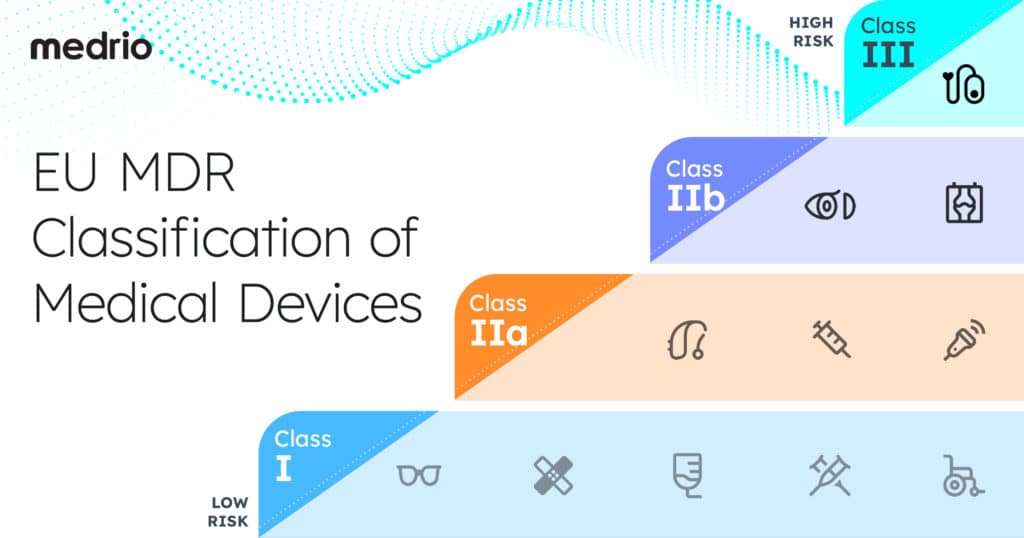
Classification of Medical Devices
The EU MDR also introduces a new system for device classification. The classification system, represented in MDR Article 51, is based on the risk posed by the device and is divided into four categories:
- Class I
- Class IIa
- Class IIb
- Class III
Manufacturers must categorize their devices by risk level and follow specific rules based on the device’s classification.
Transition Period and Compliance Deadlines
Because of the COVID-19 pandemic, medical device companies received a welcome reprieve from implementing EU MDR’s requirements. However, don’t let extensions cause you to procrastinate.
The EU MDR has instilled a sense of uncertainty within the industry, potentially slowing down research and development. These impacts mean a call to action for today, not for tomorrow.
The European Commission urges you to “…take advantage of the delay by making absolutely sure that you will be ready for the new deadline.”
Transition Period Deadlines
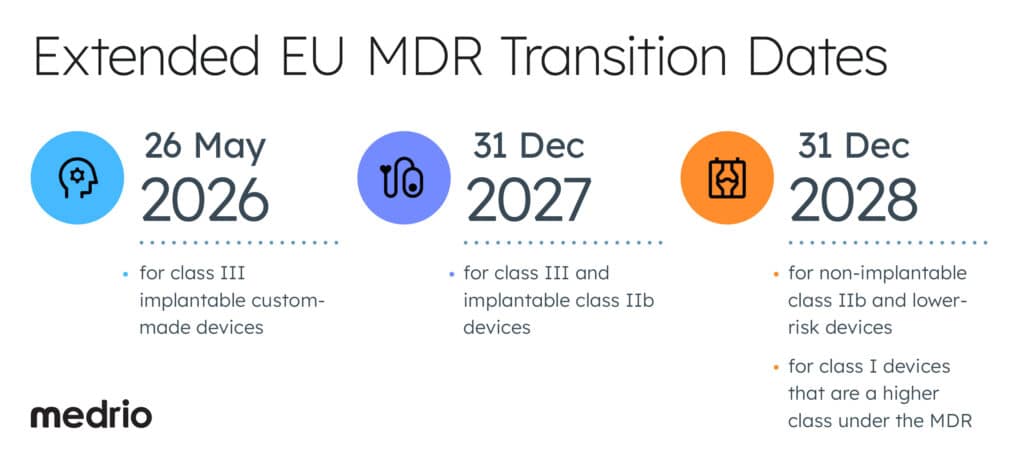
On 15 March 2023, the European Union extended the EU MDR transition periods for devices from 26 May 2024 to:
- 26 May 2026 for class III implantable custom-made devices
- 31 December 2027 for class III and implantable class IIb devices
- 31 December 2028 for non-implantable class IIb and lower-risk devices
- 31 December 2028 for class I devices that are a higher class under the MDR
EU MDR compliance is a big undertaking, and dates have shifted in the past. It’s critical to identify a key person to take ownership and stay updated with any changes.
Impacts of the European MDR
The EU MDR aims to enhance the safety of medical devices within the European Union. However, potential roadblocks have been identified in its extensive documentation requirements and the limited bandwidth of accredited notified bodies.
According to an April 2022 MedTech survey, MDR certificates have not been issued yet for more than 85% of the over 500,000 devices previously certified under the MDD or AIMDD. With so many devices requiring certification, there is currently an enormous demand for notified bodies. When it comes to clinical trials, Deborah Ann Schuster, speaking at Outsourcing In Clinical Trials: Medical Devices Europe 2023, noted that to begin investigator-initiated trials these regulations make submitting technical documentation more challenging and require substantial manpower and funding for compliance.
The Role of Notified Bodies
Notified Bodies play a crucial role in the EU MDR framework. The European Commission officially recognizes Notified Bodies as third-party organizations to assess and certify medical devices.
These organizations verify if a device meets all the regulation’s requirements before it can be used in Europe. Manufacturers must apply for a CE Mark from a Notified Body to have their device in the EU.

A Roadmap to Prepare for EU MDR
While there may be disagreement about further extensions, most experts agree on one thing: there’s a lot to be done to get ready. Although it might be tempting to take a “wait and see” approach, swift action is required to be prepared.
Here’s what to do next:
- Undertake a readiness review
- Connect with notified bodies
- Review your business case
- Brace for the potential financial impact
- Understand how EU MDR may affect future clinical research
For an in-depth, step-by-step breakdown of each of these actions and their respective implications, explore our free guide.
Undertake a readiness review
As a medical device company, you should immediately undertake a readiness review. By doing so, you’ll know what changes to make and if you want to keep investing in product development.
Manufacturers should also contract with a notified body as soon as possible for a conformity assessment before a certificate’s expiry. Doing so means that if a certificate expires prior to the enforcement of the proposed amendment, its validity can be extended.
Discover the specific steps for your readiness review, like reviewing existing certificates and more, in this guide.
Connect with notified bodies
The lack of capacity amongst notified bodies creates a huge bottleneck. As demand outstrips availability, the pressure will simply increase until more private companies are certified to act as notified bodies.
So, it’s important to build and keep relationships with preferred notified bodies and testing houses before the demand increases.
Review your business case
Device makers need to assess the costs of achieving conformity and then look at bottom-line numbers. Is it worth continuing with a device? Is it time to re-invest? Or divest?
The EU MDR is clear about its conformity requirements. Medical device manufacturers have the information required to reassess their device’s business case and decide how to move forward.
For an easy checklist of what to review, explore this guide.
Brace for the potential financial impact
Device companies may face higher costs to comply with the EU MDR. For example, the cost of one specific catheter increased 10-fold under the new rules.
To understand the cost implications for your devices, take a financial forecast of costs associated with continuing under the EU MDR. If you desire to go forward but need to make many changes, it may be time to seek more funding.
Understand the effects on future clinical research
The uncertainty around EU MDR may, at least temporarily, curb innovation, as some device companies adopt a “wait and see” approach. However, each delay from device companies may pause medical device research initiatives and investments.
The EU MDR may also affect how researchers write and present future trials. Take post-market surveillance, for example. The EU MDR requires manufacturers to actively gather, record, and analyze relevant data on device quality, performance, and safety throughout its lifetime.
The Future Under the EU MDR Regulation
The European Medical Device Regulation represents a significant shift in the regulatory landscape for medical devices in the EU. It sets stricter rules for medical device manufacturers.
The new regulation may be challenging. However, its purpose is to enhance the safety and quality of medical devices in the EU. Medical device companies must start planning for full compliance as the transitional periods come to an end.
Keeping track of all the necessary steps to prepare for EU MDR is no small feat. With expert-backed insights and in-depth research, explore our guide to help you prepare for what’s next.
Note: The information in this document is not comprehensive and is comprised of Medrio’s views. It does not constitute legal or other professional guidance. Please refer to proper industry documentation and consult with your own advisor or known notified bodies for legal or professional advice.