
Contributing Expert at Medrio: Tina Caruana, Director of eClinical Solutions
In May 2024, the FDA Laboratory Developed Tests (LDTs) final rule was released.
According to the FDA, this new policy will help ensure the safety and efficacy of these tests, while assuring patient access and reliance. Meanwhile, many industry leaders oppose the new policy.
In July 2024, the House Appropriations Committee told the FDA to suspend the final rule’s implementation and partner with Congress to revise it.
In this article, you will learn:
- Definitions & examples of Laboratory Developed Tests (LDTs)
- Main takeaways from the FDA LDT regulation
- Enforcement timelines
- Industry reactions
- FAQs
Medrio has supported more than 900 diagnostics clinical trials. Connect with our experts to get the support you need.
What is a Laboratory Developed Test (LDT)?
LDTs are a category of in vitro diagnostics (IVDs) developed by a specific laboratory for use within that laboratory. Until recently, they were exempt from FDA oversight.
LDTs may be used to measure or detect substances or analytes in the body, such as proteins, glucose, cholesterol, or DNA. This information can be used to diagnose, monitor, or determine treatment for diseases and conditions.
Examples of laboratory developed tests include:
- Toxicology panels to identify drug exposure
- Toxicology panels to monitor treatment compliance
- Newborn screening for early diagnosis of serious conditions
- Panels to monitor organ rejection after transplant
- DNA testing to detect cancer-associated mutations
- Diagnostic tests for rare diseases
LDTs are often developed out of necessity because no test is commercially available. In some cases, available assays don’t meet the unique needs of a specific patient population.
The FDA estimates that 80,000 LDTs are currently on the market, with nearly 8,000 new LDTs introduced each year.

Laboratory developed test (LDT) vs In vitro diagnostic (IVD)
Typically, IVDs are produced by device manufacturers. Some, however, are produced within a laboratory for use within that same laboratory. This specific type of IVD is called a laboratory developed test (LDT).
Until now, LDTs were under the jurisdiction of the Centers for Medicare and Medicaid Services (CMS) and exempt from FDA review. IVDs developed by device manufacturers, however, were subject to FDA review.
In the current FDA’s final rule, LDTs will be held to the same review process by the FDA as all other IVDs.
Main Takeaways from the FDA LDT Regulation
Until now, the FDA has taken an enforcement discretion approach to LDTs. They are phasing out this hands-off approach.
In the final rule, the FDA makes it clear that IVDs are devices under the Federal Food, Drug and Cosmetic Act, including when the IVD manufacturer is a laboratory. IVDs manufactured by laboratories will now fall under the same enforcement approach as other IVDs.
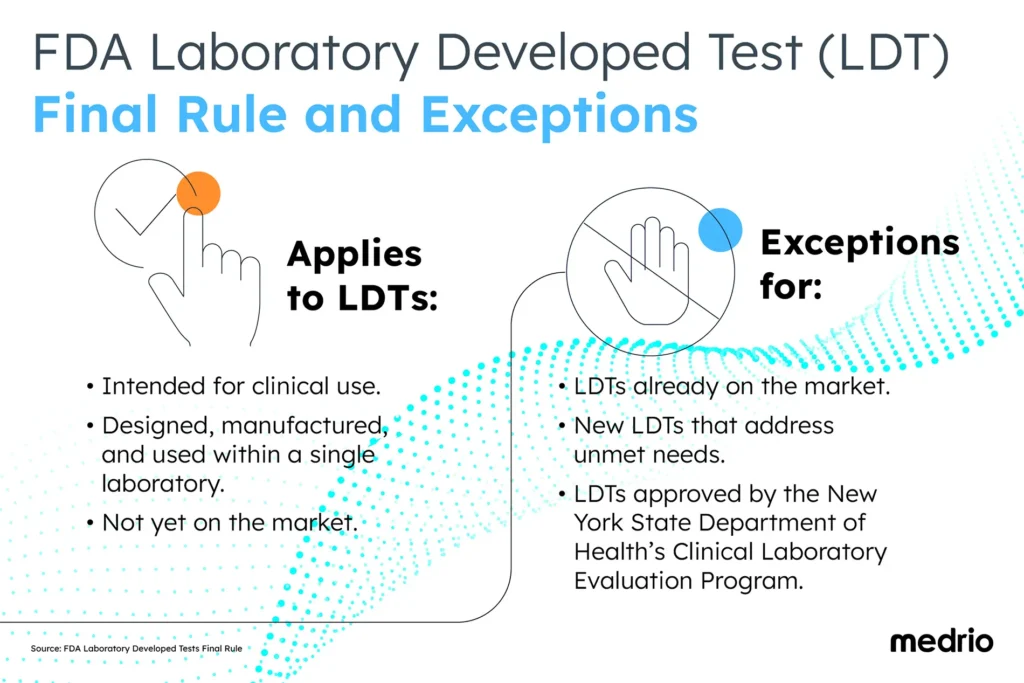
The final rule applies to LDTs:
- Intended for clinical use.
- Designed, manufactured, and used within a single laboratory.
- Not yet on the market.
The FDA states that “the risks associated with most modern LDTs are much greater than the risks that were associated with LDTs used decades ago.”
The agency expressed concern that some LDTs may not provide accurate test results. It also casts doubt on whether LDTs perform as well as FDA-authorized tests and others complying with FDA requirements.
The FDA cites data from the Centers for Disease Control and Prevention (CDC), stating that “70% of today’s medical decisions depend on laboratory test results. Given the role these tests play in modern medical care, their accuracy and validity have a significant impact on public health.”
There are a few exceptions to the final rule, including:
- LDTs that are already on the market.
- New LDTs designed to address unmet needs will remain under the former regulatory framework.
- LDTs approved by the New York State Department of Health’s Clinical Laboratory Evaluation Program are excluded from premarket review requirements.
What does this mean for LDT manufacturers?
Under the FDA’s new policy, LDTs are now subject to a more rigorous premarket review process. Manufacturers must register new LDTs with the FDA and report adverse events to the agency.
LDTs will now fall under device requirements, including:
- Adverse event reporting
- Labeling requirements
- Investigational use requirements
- Quality system requirements
- Premarket review
Phaseout policy enforcement: Laboratory Developed Tests regulation
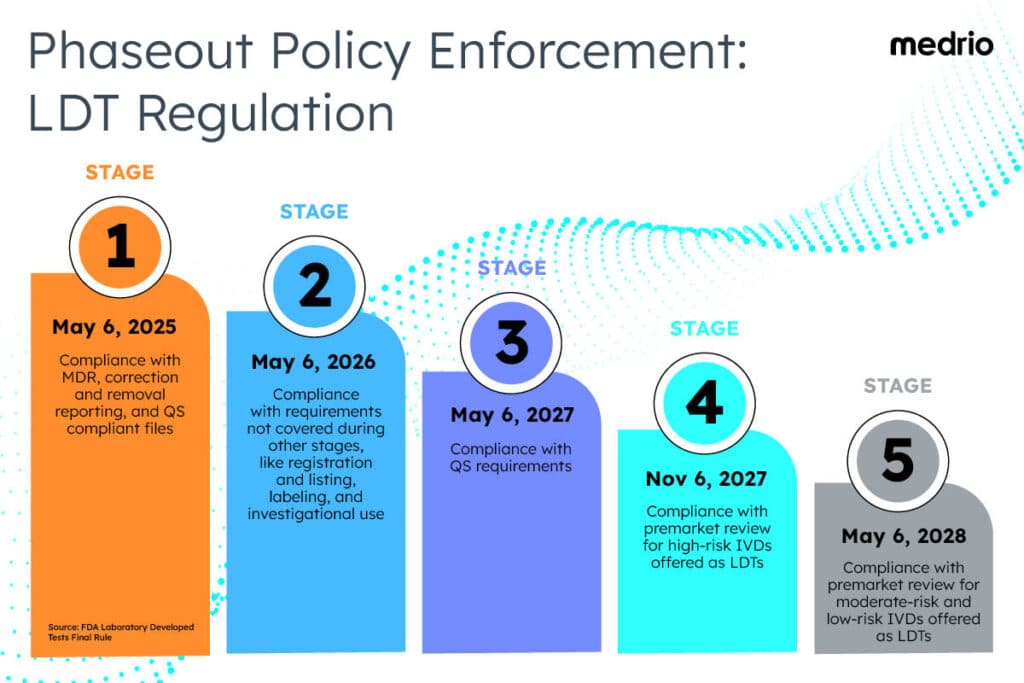
Under the final rule, the FDA’s former approach to LDTs will be phased out over four years to address patient access and reliance.
Stage 1: Beginning on May 6, 2025, the FDA expects compliance with medical device reporting (MDR) requirements, correction and removal reporting requirements, and quality system (QS) requirements regarding complaint files.
Stage 2: Beginning on May 6, 2026, the FDA expects compliance with requirements not covered during other stages of the phaseout policy, such as registration and listing requirements, labeling requirements, and investigational use requirements.
Stage 3: Beginning on May 6, 2027, the FDA expects compliance with QS requirements.
Stage 4: Beginning on November 6, 2027, the FDA expects compliance with premarket review requirements for high-risk IVDs offered as LDTs. If the FDA has already received a premarket submission by the beginning of this stage, then it will continue to exercise enforcement discretion during its review.
Stage 5: Beginning on May 6, 2028, the FDA expects compliance with premarket review requirements for moderate-risk and low-risk IVDs offered as LDTs that require premarket submissions. If the FDA has already received a premarket submission by the beginning of this stage, it will continue to exercise enforcement discretion during its review.
Following this phaseout, the FDA expects LDT makers to meet all requirements. An exception will be made if laboratories can leverage certain requirements under the Clinical Laboratory Improvement Amendments (CLIA).
Industry Reaction to the Final Rule
Overall, the final rule has not been well-received by the industry. Some experts have raised concerns that the new regulations are problematic.
Industry concerns include:
- Delays or prevention of patient access to innovative diagnostics
- Reduced patient access to testing
- Barriers to rare disease testing
- Slowed testing response to health crises
- Prohibitive costs incurred by LDT manufacturers to meet FDA approval
In May 2024, the American Clinical Laboratory Association (ACLA) sued the FDA in an effort to stop the FDA’s new LDT rule.
The ACLA argues that LDTs are services carried out by laboratory professionals. They argue that the FDA does not have the authority to enforce stricter standards on LDTs, nor regulate them as medical devices, and accuses the agency of regulatory overreach. The ACLA insists that LDTs are federally regulated by CMS under CLIA.
In July 2024, the House Appropriations Committee expressed concern about implementing the final rule, stating that it has “the risk of greatly altering the United States’ laboratory testing infrastructure.” The final rule’s implementation has been halted until the FDA partners with Congress to revise it.
The Future of LDT Testing
Although the House Appropriations Committee has temporarily halted the final rule’s implementation, labs making LDTs must now clearly understand what these requirements may entail.
The FDA promises to hold educational webinars, publish guidance documents, provide templates, and participate in conferences to help labs navigate the transition. Any IVD manufacturer can also use the FDA’s question submission process for answers to specific questions.
Medrio has supported more than 900 diagnostics clinical trials. Connect with our experts to get the support you need.
FAQs About the FDA’s Final Rule for LDTs
What is the difference between a LDT and an IVD?
LDTs are a category of in vitro diagnostics (IVDs) developed by a specific laboratory for use within that laboratory.
What qualifies as a laboratory-developed test?
Laboratory developed tests are IVDs developed for clinical use by a specific laboratory for use within that laboratory. IVDs are used in the collection, preparation and examination of specimens taken from the human body, such as blood, saliva or tissue. IVDs, including LDTs, are used to measure or detect substances or analytes in the body such as proteins, glucose, cholesterol, or DNA. This information can be used to diagnose, monitor, or determine treatment for diseases and conditions.
Does the FDA regulate laboratory-developed tests?
Since the final rule for Laboratory Developed Tests (LDTs) was released in 2024, the FDA will gradually phase in increasing regulatory requirements over the next four years.
Why did the FDA change its policy regarding LDTs?
The FDA states that the final rule is intended to “better protect the public health by helping to assure the safety and effectiveness of IVDs offered as LDTs, while also accounting for other important public health considerations such as patient access and reliance.” It also states these changes will help foster innovation within IVDs.
Does the FDA’s LDT final rule apply to all LDTs currently in use?
No, the FDA intends to exercise “enforcement discretion” for LDTs that were first marketed before the final rule was issued in May 2024.
What will happen to LDTs that fill an unmet need?
The FDA included an enforcement discretion policy for LDTs manufactured and performed by laboratories integrated within a healthcare system if no FDA-authorized IVD is available that meets the patient’s needs.
Is there an enforcement discretion policy that applies to modifications of currently marketed IVDs offered as LDTs?
The FDA indicates it will generally not enforce premarket review and QS requirements for modifications to LDTs marketed prior to the date of issuance of the rule, except for these changes:
– Change the indications for the use of the IVD.
– Alter the operating principle of the IVD.
– Include significantly different technology in the IVD, such as the addition of AI.
– Unfavorably change the IVD’s performance or safety specifications.